
全球健康药物研发中心郭晋疆:多元科学计算系统在药物研发管线中的搭建与实践

摘要:2022 年 8 月 5 日,2022 阿里云生命科学与智能计算峰会在北京望京昆泰酒店举行,全球健康药物研发中心数据科学部负责人郭晋疆博士,带来了题为《多元科学计算系统在药物研发管线中的搭建与实践》的分享,以下是他的演讲内容整理,供大家阅览:

全球健康药物研发中心数据科学部负责人 郭晋疆
01 科学计算驱动药物研发的趋势
下图摘自 2022 年初的 Nature Reviews,可以看到以科学计算或人工智能驱动的药物研发项目由 2010 年的 6 个增至 2021 年的 158 个, 11 年增长超 28 倍。而传统药物研发项目从 705 个降至 333 个,虽然它依然是主要的药物研发模式,但已呈现下降趋势。
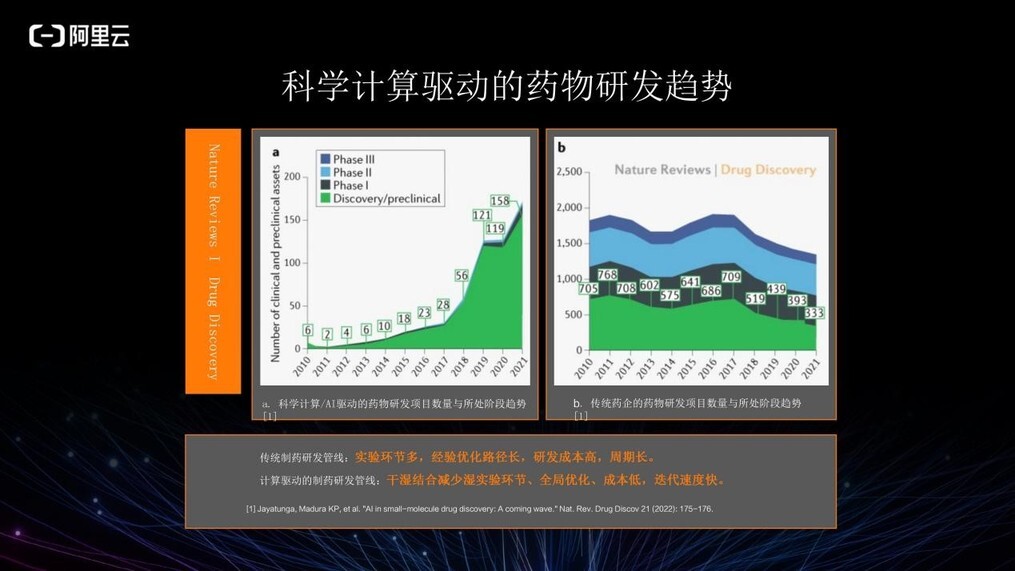
传统药物研发管线需要涉及大量湿实验环节,且多数基于科学家的个人经验和实验结果来进行优化,优化路径长,研发成本高昂,周期也长。与之形成对比的是以计算驱动的药物研发管线,它是一种干湿结合的形式,减少了湿实验环节。并且很多数据驱动的方法学习了历史或全球范围内的实验数据,在优化化合物时更倾向于全局的优化,优化过程更快,成本更低,迭代速度也更快。
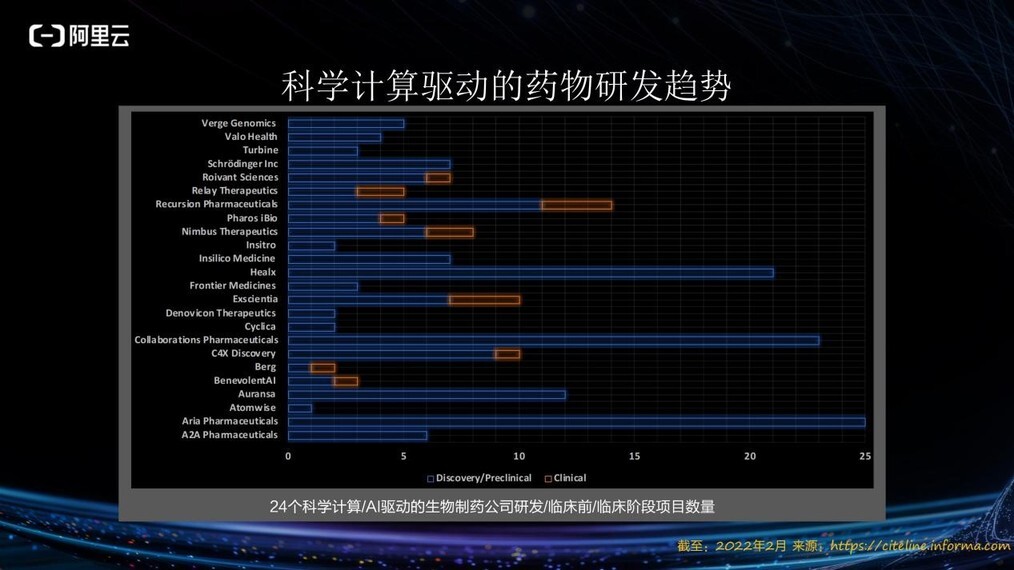
上图为全球 24 家以科学计算/AI 驱动的生物制药公司在研药物情况,其中有 15 款计算驱动的药物已经进入临床实验阶段。相信在不久的将来,会有更多计算驱动的药物成功上市,惠及更多病患。
02 药物研发管线不同阶段的特质与问题
全球健康药物研发中心作为创新型的小分子药物研发机构,也在使用多种计算方法解决药物研发早期阶段不同的问题。

药物研发早期阶段的一般流程如下:
Stage 1:疾病生物学,即疾病的确立。疾病可以粗略地分为外源性疾病和内源性疾病,其中外源性疾病指外来生物体或非生物体侵入人体造成的一些组织性病变,比如有害微生物、病菌、病毒、疟原虫或粉尘等非生物体;内源性疾病指人体基因变异或机能失调造成的组织性病变,比如各类肿瘤、心脑血管疾病、慢性病和罕见病。

全球健康药物研发中心聚焦于全球健康的公共领域,我们不仅关注外源性的传染性疾病比如结核病、冠状病毒、疟疾和寄生虫感染,也关注内源性疾病,比如一些肠道类疾病 EED 等。
Stage 2:靶标确立与验证,即与疾病强相关的蛋白或生物标记物。此阶段会面临纷繁多样的异质化数据,研究人员需要分析疾病机制、疾病在生物网络通路中的表现,也会包含一些基因变异以及表达等多组学信息。
Stage 3:苗头化合物确立,筛选或设计能够与蛋白产生相互作用的分子,即苗头化合物。目标是一方面在分子化合库中筛选可能产生活性的小分子,另一方面也需要设计创新型的活性分子。该阶段存在并可以获得大量实体或虚拟的化合物库数据,数量可达亿级,例如 Chemdiv, Zinc 等。但是针对靶标蛋白的活性化合物分子比较稀少,尤其是一些罕见性疾病或人类不是特别关注的疾病。
Stage 4:苗头-先导化合物优化
Stage 5:临床前候选药物。这 2 个阶段需要考量的不仅仅是化合物与靶标蛋白的相互作用,也要综合药代动力学、合成工艺、可成药性,如分布、代谢、毒理等,在平衡各项性质之后,优化设计出一种真正有效且安全的药物。这是一个综合优化的过程,涉及到大量 ADMET 数据的收集以及训练建模。与此同时,也会有少量研发管线项目的实验数据。
03 多元科学计算系统的构建
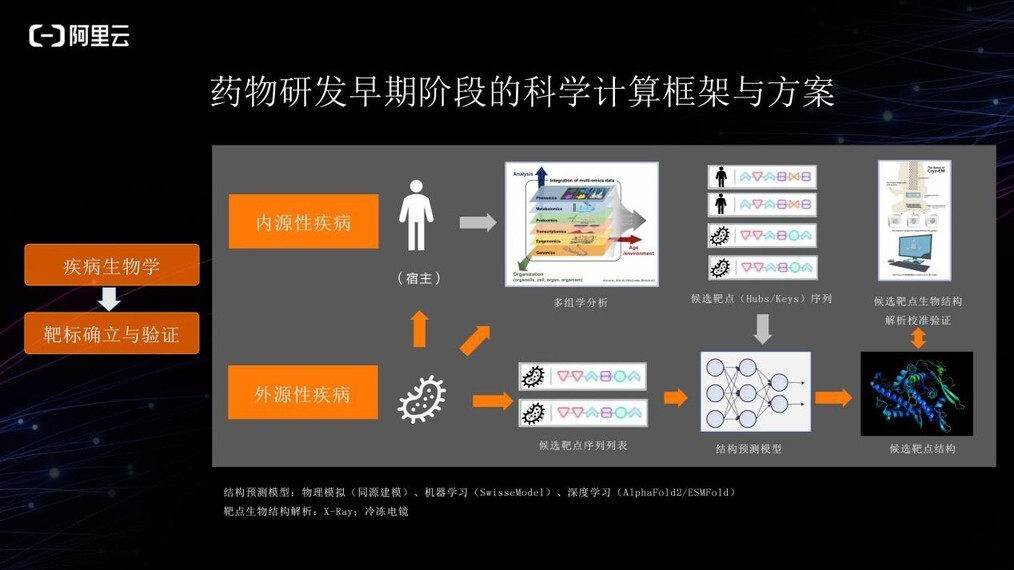
疾病的选择到靶标确立阶段,数据纷繁多样且异质化。
针对内源性疾病,通常会进行多组学的分析。通过分析正常人和病患的代谢组学、基因组学或蛋白组学等多组学信息,找到与疾病强相关的 Hub 基因/蛋白或关键基因/蛋白,作为靶标的候选。得到蛋白序列之后,使用结构预测模型预测其 3D 结构。结构预测模型中,Alphafold 是近年的创新型深度学习方法,还有此前的传统机器学习、物理建模等方法也可以得到候选靶标结构。
针对外源性疾病,找到靶标的方法包括:
1、可以通过分析人体免疫机制,比如融合机制来研究人体的多组学信息,找到人的关键性靶标;
2、也可以直接分析菌落的多组学情况,确定关键通路中的蛋白作为靶标的候选;
3、对于一些相对比较简单的病原体,例如病毒,可以直接获取它在侵入人体融合或转录过程中的蛋白序列,进行所有相关蛋白结构预测并提供给生物学家或化学家分析,用于确定靶标。
结构生物学将需要确定的蛋白的真实结构进行解析、并对预测的 3D 结构验证与校准,以便后续阶段的分析与预测。
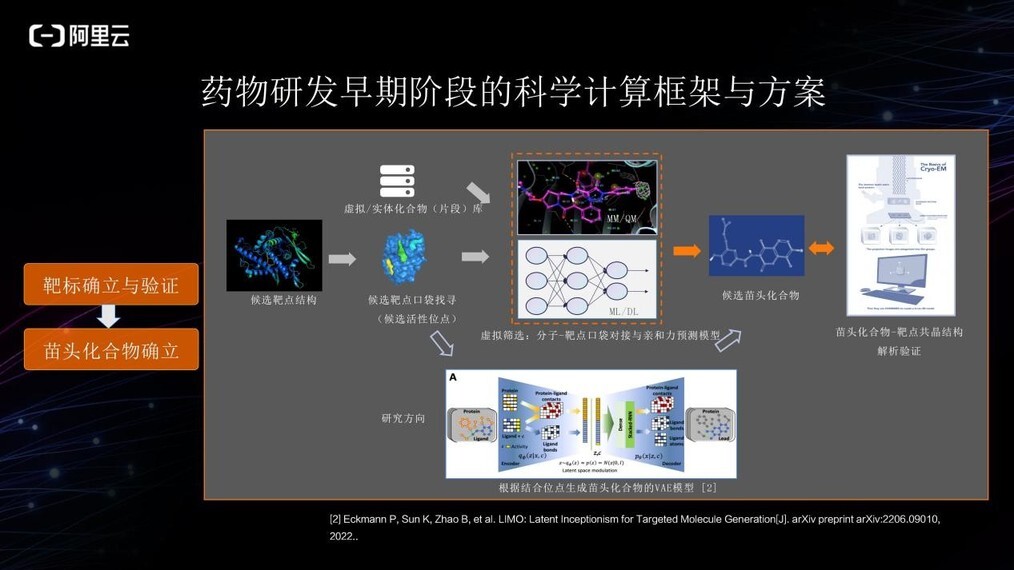
确定靶点蛋白后,接下来需要在靶点上找到可能的与分子结合的口袋,结合口袋指分子化合物可以与之产生相互作用的结合位点。通过计算来判断化合物能否与靶标产生相互作用,即是否有潜在活性,主要有两大类计算方法:
1、使用分子力学或量子力学等物理学模拟的方法;
2、使用机器学习或深度学习的方法。利用这两类方法在已知/虚拟生成化合物库中虚拟筛选出与靶标相互作用可能性较高的化合物,作为候选苗头化合物。
除了使用虚拟筛选化合物库的方式, 越来越多的研究人员试图采用端到端的方式从口袋理化性质直接设计苗头化合物,这样可以跳过物理模拟或机器学习虚拟筛选化合物库的部分,用 AI 直接生成有潜在活性的苗头化合物,相信这也会成为未来的重点研究方向之一。
获取到候选苗头化合物之后,将由生物、化学方面的专家进行湿实验验证或者结构生物学进行化合物-靶点共晶结构的解析验证,确认其是否符合预测的结果,并用于下阶段的化合物优化。
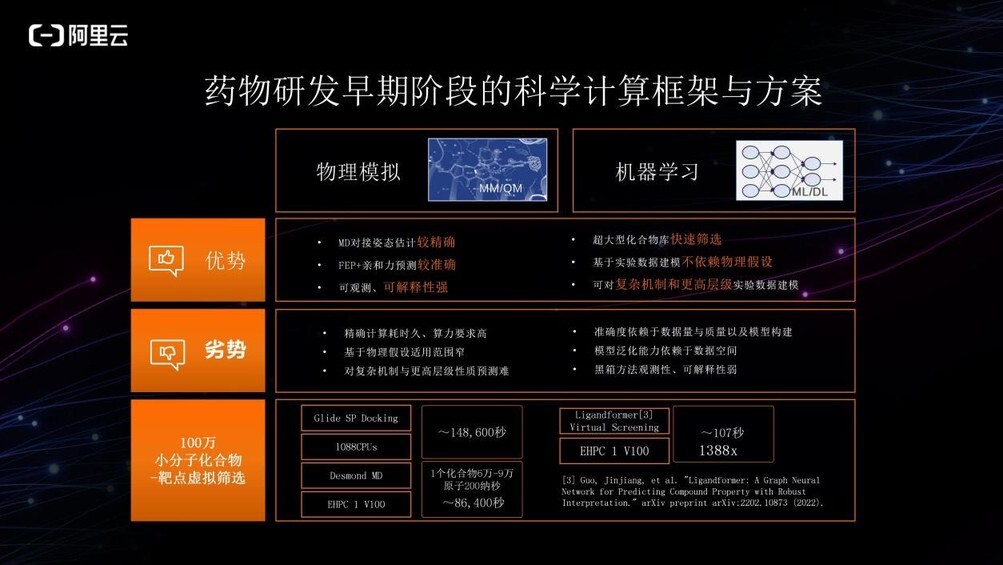
对物理模拟与机器学习方法进行比较,物理模拟是目前很多药企较常使用的一类方方法,其优势在于 MD 对接姿态估计较为精确,FEP+亲和力预测较准确。且采用 3D 建模,可以直观地看到小分子和口袋的结合情况,可解释性也较强;而劣势在于需要的算力非常高,有弹性超算的需求。此外,它基于物理假设,能够适用的范围较窄,无法应对一些复杂的机制,比如多靶点或蛋白变构现象的预测,或更高层级的比如化合物在细胞、类器官或人体组织层级的性质预测等。
机器学习方法主要通过已知数据训练优化给定数学模型的参数,因而经过数据训练产生的模型大小是固定的,可用模型快速筛选超大型的化合物库。其次,它基于经验数据或实验数据,不依赖于物理假设,能够应对复杂机制或更高层级的性质进行数学建模和预测;其劣势在于它很大程度上依赖于数据的质量以及数据空间的分布情况。数据储量大质量高,则机器学习或深度学习的表现好,反之则可能表现较差。此外,其泛化能力也非常受限于它能够看到的数据空间,而且机器学习是一种黑箱方法,科学家很难明确其判断依据。
以虚拟筛选 100 万个小分子化合物为例,使用物理模拟方法 docking 大概需要 148,600 秒,而在 v100 的 GPU 上使用深度学习方法只需 107 秒,速度相差 1000 多倍。另外,通过精度更高的分子动力学方法在机器上模拟一个化合物与靶标蛋白位点的结合,在 6 万-9 万原子体系中模拟 200 纳秒时长,在 v100 的 GPU 上大概需要 86,400 秒,由此可见基于物理模拟的方法要求很高的算力。
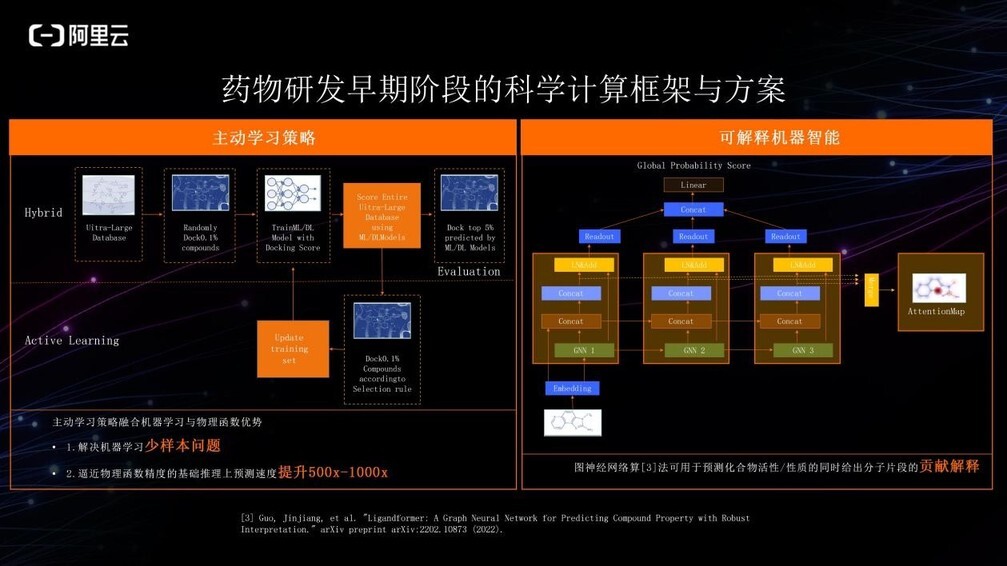
在早期苗头化合物的发现和确立过程中,研究人员通常能够获得针对靶点的实验数据非常少。如果直接用这些数据来做深度学习算法的建模,机器只能看到非常有限的化学空间,训练出的模型的泛化能力与预测鲁棒性较差,因此我们采用了主动学习的方式,使用专家经验或一些物理函数校准 AI 模型,不停地增广训练集,迭代几轮之后模型即可投入使用。
此外, 由于很多 AI 模型本身是黑箱模型,生物学家或化学家可能无法完全信任它给出的结果。为此我们自研了基于 self-attention 机制的深度学习算法 Ligandformer,模型能够在给出化合物的性质或活性预测分值的同时,也会给出分子片段对活性/性质的贡献解释,供科研人员参考和借鉴。
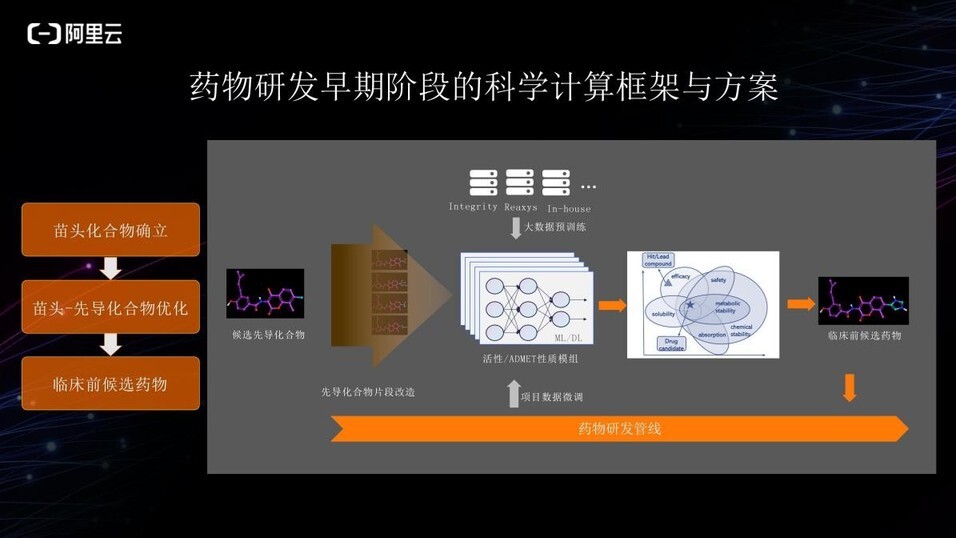
从苗头化合物到先导化合物,再到临床前的候选药物过程中,需要将苗头化合物进行一系列优化改造。优化过程中,计算层面一般流程是采用大数据对不同性质的模型进行预训练,得到 pretrained model,并通过实际研发管线中的实验数据对 pretrained model 进行微调,然后用微调后的模型大批量筛选各种改造的先导化合物结构。最终在平衡多种性质之后,得到候选药物列表,提供给生物学家或化学家参考选择并进行下一步的湿实验验证。
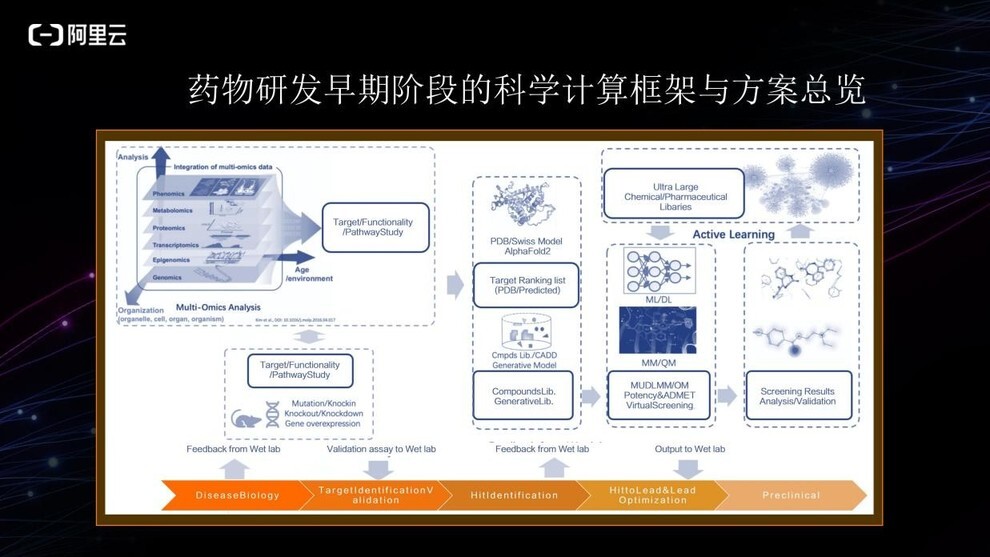
上图可见,计算过程贯穿整个药物研发的早期阶段。
04 多元科学计算系统 E-HPC 平台实践

2020 年新冠肺炎爆发初期,阿里云团队与我们合作搭建了抗击新冠肺炎的公共信息平台,搜罗来自全球信息源的关于病毒研究。与此同时,我们也搭建了预测性的服务平台,这是在超算平台上搭建的对外服务,免费开放给科学家们使用。目前已对服务进行升级和优化,在 20 余个内外部合作项目中广泛使用。
此外,我们收集整理了大量来自全世界商业和非商业数据库的数据,建立了可视化结构-性质数据分析工具,帮助科学家更好地进行研究。
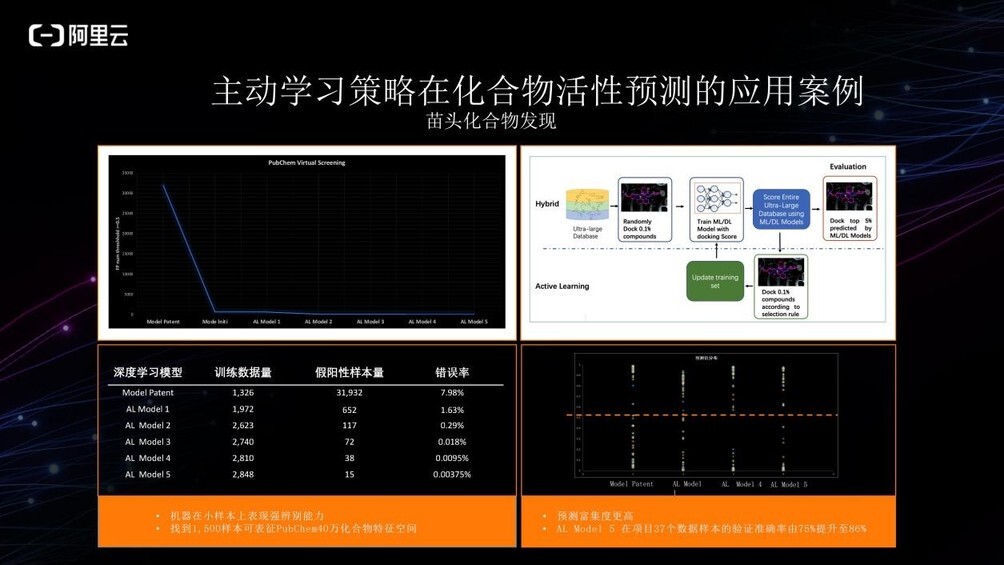
在此前的一次苗头化合物发现的项目中,当时我们需要将 PubChem40 万化合物库的化学空间较好地进行表征与筛选。我们使用主动学习策略训练深度学习模型并筛选化合物库,在主动学习进行 5 轮迭代后,错误率由最初的 7.98%下降到了不足万分之一。与此同时,依靠专家经验不停地增广训练数据样本,训练数据样本仅增加了 1500 余个。总共 2800 多个的训练数据量并不是很大,但它使得机器学习模型表现出比较强的辨别能力,可以辨别 40 万化合物库中化学空间的情况。
同时,我们对项目中 37 个实验数据进行回溯性验证。从最初的模型到第五个模型,准确率由 75%提升至 86%。

我们与北京协和医院进行了罕见病相关研究工作,使用了内部自研的生物信息网络相互作用关系算法来重新校准蛋白-蛋白相互作用网络。通过校准后的网络再综合生物信息统计学方法找到了 ATTR 罕见病的新药物,与此同时也重新定位到了一款淋巴性白血病的药物。这项工作已被某医学期刊收录。

总的来说,基于分子力学的方法主要应用于已知靶点或需要确定靶点的任务上,比如早期的靶点确立、苗头化合物确立与苗头-先导化合物优化阶段;基于机器学习/深度学习的方法可以应用于 苗头化合物确立、苗头-先导化合物优化阶段以及临床前候选药物优化阶段,除此之外还可应用于未知靶点的场景,比如只有一些表型数据需要通过数据驱动建模,比如药物研发后期对细胞、组织、类器官或人体层级性质的预测、可成药性分析等。
05 挑战与机遇

未来,我们将在以下几个方面进行深入研究:
第一,复杂治病机制和靶点研究。比如细菌的耐药性研究、蛋白变构现象的预测等。
第二,靶点活性位点的突变预测。比如冠状病毒会持续变异,药物在变异的位点的有效性,可以通过计算分析判断。
第三,创新药物的分子设计。越来越多的研究人员聚焦在基于蛋白靶点口袋的活性分子进行生成和设计,同样也可以基于表型数据端到端地生成和设计分子化合物。
那么,如何解决或突破问题?首先,数据必不可少。除了分子化合物的理化性质的数据以外,可以将更多的横向数据比如生物信息网络或通路中的网络信息数据融合进来,也可以将更低层级的数据比如电子云密度数据融合进来。
而庞杂、多元化、异质化的数据需要强有力的算法能够融合不同层级、不同尺度的数据,并且能够在数据上提取出模式特征做最终任务的预测。而这一切必然离不开超算平台,因此我们对于超算平台的需求也逐渐增大,我们需要有更大规模的数据承载以及处理能力,需要有更快的速度与进度。
相信结合数据、算法和超算平台的通力合作,再加上跨专业、跨行业领域人才的共同努力下,药物研究行业即将取得更大的突破。
点击这里,观看嘉宾在本次峰会的精彩演讲视频。
版权声明: 本文为 InfoQ 作者【阿里云弹性计算】的原创文章。
原文链接:【http://xie.infoq.cn/article/a4292b37e87818f722d8c3b81】。文章转载请联系作者。

澎湃算力,无处不在。 2018.08.24 加入
阿里云弹性计算团队,关注虚拟化、通用计算、异构计算以及云上HPC和云上运维CloudOps。







评论